I thought now would be a good time to review some of the research put out by World Community Grid in the past as WCG projects and volunteers have done and continue to do a lot of valuable work.
In 2007, the authors Max W. Chang, William Lindstrom, Arthur J. Olson, and Richard K. Belew at UCSD published a paper entitled "Analysis of HIV wild-type and mutant structures via in silico docking against diverse ligand libraries".
HIV (Human Immunodeficiency Virus) causes the debilitating disease AIDS (Acquired Immune Deficiency Syndrome) which results in the slow destruction of one's immune system, thereby predisposing individuals to a wide variety of other infections. HIV/AIDS is a major public health hazard and is considered to be a a global pandemic. Especially hard hit areas include sub-Saharan Africa where 66% of all AIDS deaths occur.. The HIV virus itself is an RNA virus within the retroviridae family. Such viruses are known to integrate themselves into the genomes of their hosts via the enzymes reverse transcriptase and integrase.
The paper arose from a FightAIDS@Home distributed computing project that used a BOINC program called AutoDock to screen HIV protease structures against a panel of 1771 ligands (molecules that potentially bind to and stick to the HIV protease). The HIV protease refers to a protein found within the HIV virus that chops up larger proteins (think a set of scissors that cuts only protein) into smaller components that are required for the virus to mature. The FightAIDS@Home study included both already known HIV protease inhibitors as well as other molecules not previously known to act as protease inhibitors.

Credit - Nupar, member of World Community Grid and wikipedia
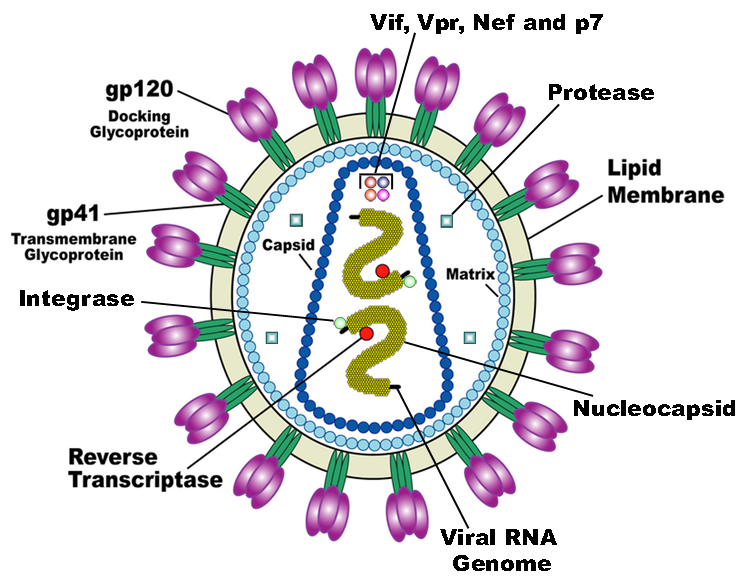
Credit- US National Institute of Health
The HIV protease in particular cleaves larger proteins (known as a polyprotein) into smaller component parts, such as the individual capsid, nucleocapsid, matrix, integrase, and reverse transcriptase proteins. Thus, it is an essential component required for the next generation of viruses to mature successfully. As such, the protease is a major therapeutic target for anti-HIV drugs.
One of the main focuses of the article (and FightAIDS@Home) therefore was to determine what compounds/ligands might be able to function as protease inhibitors for potential therapeutic purposes. In addition, the authors sought to determine what HIV protease structure would be representative of the wildtype (or circulating versions) of the HIV protease as a whole. In essence, they wanted to find an ideal protease structure that could be found to capture the central tendency of the entire set, and if so, this single probe could be used as an initial probe against large libraries of ligands. They determined through calculation of binding energy profiles of the PDB structures that the protease structure 2BPZ best captured the central tendency of the HIV proteases. Thus, this structure could be used in novel virtual screens of HIV protease inhibitors.
The paper was cited by 10 additional papers since 2007 so has certainly not gone unnoticed.
With additional computation thanks to the likes of boinc volunteers and team gridcoin more science will continue to be churned out!