3.0 The BH3-only proteins: The direct activators
3.1 BID
The BH3-only protein BID resides mainly in the cytosol, and after activation it translocates to the OMM where it plays an important role in BAK and BAX activation, and MOMP.BID is predominantly activated via the extrinsic death pathway. In response to pro-apoptotic stimuli BID is cleaved by caspase-8 via death receptors, resulting in tBID. BID can also be cleaved by other proteases such as caspase-2 and granzyme B. BID has multiple cleavage sites for different proteases. Proteolytic cleavage of BID by caspase-8 occurs at the large unstructured loop joining the inhibitory N-terminal and the C terminal. The N terminal fragment is quickly degraded by ubiquination, whereas the BH3-containing C terminal fragment (tBID) undergoes myristolation at a glycine residue site on the newly exposed N terminal, which promotes OMM targeting. The mitochondrial carrier homologue 2/Met-induced mitochondrial protein (MTCH2/MIMP) has been shown to be a major facilitator in the recruitment of tBID to the mitochondria (Zaltsman et al., 2010: Cogliati and Scorrano, 2010). MTCH2/MIMP is an OMM surface exposed protein, and the mechanism by which it facilitates tBID translocation is still unknown. Knockout studies (Zaltsman et al., 2010) have shown that absence of MTCH2/MIMP does not completely abrogate tBID-induced apoptosis suggesting that other OMM receptors may play a role in tBID translocation.
Cardiolipin (CL) is an intergral mitochondrial membrane component. The activated tBID interacts with the OMM via CL binding. The α6 helix domain of tBID is responsible for CL targeting through electrostatic interactions of lysine 157 and 158 (Gonzalvez et al., 2010). Interaction of tBID with CL disrupts mitochondrial bioenergetics in a BH3-independent manner. CL also provides an anchor and an activating platform for caspase 8 (Gonzalvez et al., 2008). This localization of caspase 8 ensures that cleavage of BID occurs when needed.
Whether tBID interacts primarily with MTCH2/MIMP or CL for OMM insertion is unknown. MTCH2/MIMP has been found under certain stimuli to reside in a 185 kDA complex that also comprises of tBID and BAX. Inactive BID has been shown to be able to translocate to the OMM, but is unable to promote MOMP on its own. A logical sequence of translocation for tBID would involve binding to CL first via its α6 helix (where it causes the disruption of the OMM, and any BID that translocates can be truncated by caspase 8), and then binding to MTCH2/MIMP where it can associate with and activate BAX or BAK via its BH3 domain.
Cleavage of BID to tBID has been shown to occur downstream of MOMP (Shelton et al., 2009) suggesting that the main role of tBID as a direct activator in the intrinsic death pathway is to determine irreversible commitment of apoptosis by feed forward amplification by subsequent activation of the executioner caspases downstream of APAF-1.
Multiple alternatively spliced isoforms of BID have been found, but their nature is still unknown.
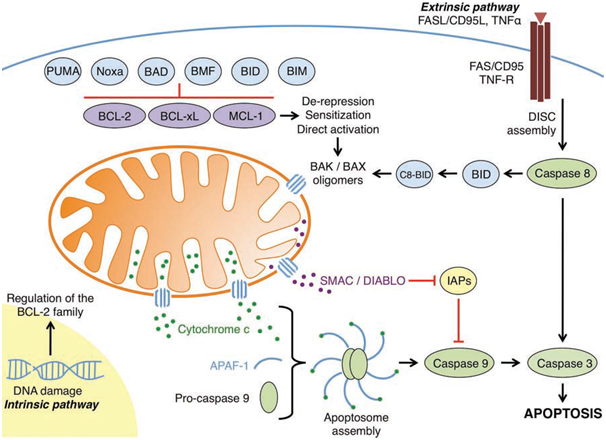
Figure 1: An overview of the intrinsic and extrinsic apoptotic pathways
(Elkholi et al., 2011).
3.2 BIM
The BH3-only protein BIM is an unstructured polypeptide with its binding affinity based solely on its BH3 domain. It is expressed as three alternatively spliced isoforms, BIM-short (BIM-S), BIM-long (BIM-L), and BIM-extra long (BIM-EL) with the latter two being the most often expressed. BIM-S is less abundant in cells, presumably due to its strong apoptotic activity. The two main isoforms of BIM, BIM-L and BIM-EL are sequestered by the microtubule-associated dynein motor complex via interaction with the dynein light chain (DLC) 1 until pro-apoptotic stimulus occurs such as cytokine withdrawal or growth factor withdrawal. BIM is regulated by both transcriptional and posttranslational mechanisms. The BIM promoter is regulated by stress-stimulated transcription factors such as the FOX (forkhead box) proteins, FOXO3A during cytokine withdrawal and activator protein 1 (AP-1). Growth factor withdrawal activates c-Jun N-terminal kinase (JNK) signalling which results in the phosphorylation of BIM releasing it from the dynein motor complex. BIM can be phosphorylated by a plethora of kinases. BIM has multiple phosphorylation sites which regulate its propapoptotic activity, and can stabilise the protein or cause its degradation (Hubner et al., 2008). An example of this is the extracellular-signal-regulated-kinases/mitogen-activated protein kinase pathway (ERK/MAPK). ERK1/2-mediated phosphorylation on Threonine-112 stabilises BIM promoting its apoptotic activity by increasing its binding affinity to the anti-apoptotic proteins and by protecting it from proteasomal degradation. Stabilised BIM interacts weakly with BAX, activating it and promoting oligomerisation (Merino et al., 2009). ERK1/2-mediated phosphorylation on serine-55/65/73 targets BIM for ubiquitination by a 26s proteasome or a 20s ubiquitin-independent proteasome (Wiggins et al., 2011), and lowers its binding affinity for the anti-apoptotic proteins. The ERK1/2 pathway is responsible for the majority of BIM-EL degradation. ERK1/2 mediated stabilisation of BIM-EL occurs via crosstalk of the JNK signalling pathway.
BIM-EL has been shown to be a protein kinase A (PKA) substrate (Moujalled et al., 2011) implicating phosphorylation of BIM-EL by PKA as a general process. PKA associates with BIM-EL mainly via a cyclic AMP (cAMP)-regulated PKA regulatory subunit-a (PRKAR1A). Interaction of BIM-EL with PRKAR1A enables phosphorylation of BIM-EL by PKA (Moujalled et al., 2011). PKA stabilizes BIM-EL via phosphorylation protecting it from ubiquitination and allowing it to transiently interact with BAX, resulting in oligomerisation and MOMP.
Another form of posttranslational modification other than phosphorylation has been shown to regulate BIM-EL. BIM-EL can be proteolytically cleaved at position D17 by caspase-3 resulting in an N-terminally truncated version of BIM-EL. Cleavage of BIM-EL results in enhanced killing kinetics, due to its increased binding affinity for BCL2 (Chen and Zhou, 2004). This shows that like BID, BIM-EL can be activated downstream of the caspase cascade, triggering a positive feedback amplification of MOMP, cytochrome c release, and caspase activation.
BIM is regulated by a complex system of phosphorylation and exerts it main pro-apoptotic function by binding with anti-apoptotic BCL-2 family members. BIM can interact with all of the pro-apoptotic BCL-2 family members. It most likely interacts with the OMM by binding to the OMM embedded anti-apoptotic proteins, which it has a high binding affinity for. As result of binding to the anti-apoptotic members, it prevents BAX and BAK from being sequestered. Also the binding of stabilized BIM to BAX is a weak interaction, allowing a transient interaction to occur, activating the BAX molecule and releasing itself immediately from it allowing BAX to undergo dimerisation. BIM then can therefore promote oligomerisation and immediately bind with anti-apoptotic BCL-2 proteins preventing inhibition of MOMP.
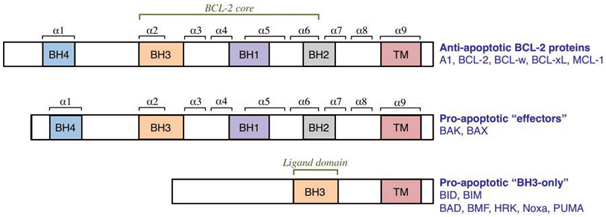
Figure 2: The three groups of the BCL-2 family (Elkholi et al., 2011).
3.3 PUMA
The BH3-only protein PUMA is normally expressed at very low levels, and accumulates rapidly in the cell after apoptotic stimulus via transcriptional induction. Multiple binding sites exist for transcription factors in the promoter region of the PUMA gene. In response to DNA damage or growth factor withdrawal, transcription of PUMA is activated by p53 (protein 53) or its homologue p73. The tumour suppressor p53 activates transcription of PUMA by binding to the PUMA promoter, modifying histones H3 and H4 through acetlylation, which opens the chromatin structure (Wang et al., 2007). Other transcription factors exist that activate PUMA in a p53-independent manner, such a FOXO3a in response to cytokine deprivation. PUMA expression is also prone to downregulation by transcriptional repressors such as Slug (Wu et al., 2005: Zhang K.,et al., 2011) preventing MOMP.
Until recently PUMA was thought to be subject to only transcriptional control. Recent evidence has shown that PUMA can be regulated via post-translational phosphorylation (Fricker et al., 2010). PUMA can be phosphorylated at multiple sites; phosphorylation at serine-10 leads to proteasomal degradation of PUMA, promoting cell survival.
PUMA has two major isoforms, PUMAα and PUMAβ which both share identical BH3 domains and similar kinetics for MOMP induction. PUMA binds with high affinity to all five anti-apoptotic BCL-2 family members via its amphipathic α-helical structured BH3 domain (Chipuk et al., 2008). Mitochondrial localisation of PUMA is directed by the hydrophobic domain of its C-terminal. PUMA primarily acts as a sensitizer and a derepressor by freeing sequestered BAK and BAX from the anti-apoptotic repertoire, and by binding to the anti-apoptotic BCL-2 proteins freeing other proteins such as BIM to activate BAK and/or BAX leading to MOMP and caspase cascade activation(Yu et al., 2008). PUMA has been shown to directly activate BAX and BAK (Chipuk et al., 2008: Garrison et al., 2011: Kimet al., 2009).
PUMA is a potent activator of apoptosis due to its binding affinity, and its ability to mediate p53-dependent apoptosis and p53-independent apoptosis. PUMA expression greatly enhances the kinetics and efficiency of MOMP. It is tightly regulated by transcription factors to protect the cell from unwarranted apoptosis, and has been shown to undergo posttranslational phosphorylation; the kinase responsible for phosphorylation of serine-10 remains to be defined. Other kinases may phosphorylate PUMA at different sites, regulating it in different ways. It is an important protein due to its potency to interact with all anti-apoptotic BCL-2 members, although much of the mechanisms involved in PUMA-dependent apoptosis remain to be elucidated.
…
The direct activator proteins all require BAK and/or BAX to cause apoptosis. The BH3 domains in tBID, BIM, and PUMA are vital as they are the main site for interaction with other BCL-2 family members. In vivo apoptosis is most likely caused by a combination of mechanisms involving all the direct activators working together in synergy. PUMA and BIM are similar in that they both are regulated by transcriptional and postranslation mechanisms. PUMA and BIM are both induced trancriptionally by many of the same factors such as FOXO3a after cytokine deprivation. This allows for greater efficacy in inducing MOMP as PUMA can interact with all of the BCL-2 anti-apoptotic family, allowing BIM to freely interact with and activate BAK and BAX.
The BH3-only protein BID is unique as it requires cleavage to drive apoptosis. After MOMP occurs by BIM or PUMA resulting in a caspase cascade, BID is likely to be activated, propagating MOMP. BIM can also be activated by proteolytic cleavage, highlighting the synergestic molecular mechanisms that drive a cell to commit to apoptosis.
@RiskDebonair
Adventure Capitalist of the Future