7.0 Cancer and the BCL-2 family
Aberrations in the homeostatic balance between cell growth and cell death can result in carcinogenesis. Alterations of protein expression in the BCL-2 family are found in many cancer types. Cancer cells with a change in protein expression of the BCL-2 family, which pushes the cell towards a survival state and prevents apoptosis; grants the cell an inherent advantage. The pro-survival protein BCl-2 is predominantly found in cancers with an unfavourable prognosis. Overexpression of this protein in cancers can give rise to resistance to therapies such as chemotherapy. In breast cancer overexpression of BCL-2 has also been shown to work in synergy with hypoxia to upregulate vascular endothelial growth factor (VEGF) resulting in tumour neoangiogenesis (Biroccio et al., 2000).
In a majority of cancers the expression of p53 is altered, commonly by a mutation in the TP53 gene. Downregulation or a dysfunctional phenotype of p53 allows the cell to bypass a crucial cell cycle checkpoint. This prevents DNA repair proteins from correcting any mutations that may have occurred, resulting in an accumulation of mutations over many cell cycles. Downregulated or dysfunctional p53 prevents transcription of many pro-apoptotic proteins (e.g. BAD, PUMA, Noxa), inhibiting the intrinsic apoptotic pathway from occurring in these mutated cells.
Although a dysfunctional regulatory apoptotic pathway may not cause cancer, aberrations in the expression of the BCL-2 effector proteins, anti-apoptotic proteins, and BH3-only proteins can contribute to the many cancer types. The BCL-2 family are important regulators of apoptosis, and it is no surprise that these proteins are being directly targeted to induce apoptosis in cancerous cells.
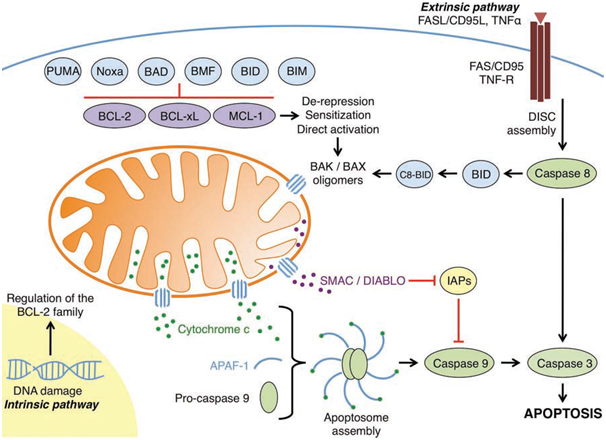
Figure 1: An overview of the intrinsic and extrinsic apoptotic pathways
(Elkholi et al., 2011).
7.1 p53-independent PUMA upregulation to induce apoptosis in cancer cells
The inhibition of protein kinases that are aberrantly activated is one of the most effective therapies to treat cancer. The mechanisms in which tumour growth is suppressed for most kinase inhibitors are unknown, as most kinase inhibitor drugs inhibit multiple kinases.
UCN-01 (a staurosporine analogue) is a non-specific potent inhibitor of protein kinases. In colon cancer cells UCN-01 has been shown to induce apoptosis via PUMA, inhibiting cancer cell growth (Dudgeon et al., 2010). UCN-01 inhibits Akt, which negatively regulates FOXO3a via phosphorylation. With Akt inhibited FOXO3a upregulates PUMA, resulting in induced apoptosis.
PUMA is a transcriptional target of p53, and in many cancers the function of p53 is abrogated. Using kinase inhibitors such as UCN-01, PUMA can be induced in a p53-independent manner in colon cancer cells. Targeting PUMA in colon cancers may serve to improve the already available therapies and to prevent chemoresistance from occurring.
In gastric cancer interferon-gamma (IFN-γ) has been shown to stimulate PUMA expression (Gao et al., 2010). IFN-γ upregulates interferon regulatory factor-1 (IRF-1); IRF-1 is a transcription factor that activates PUMA expression. IRF-1-mediated upregulation of PUMA by IFN-γ results in apoptosisin gastric cancer cells.
Both of these examples of PUMA induced apoptosis in cancerous cells highlights that PUMA can be p53-independently therapeutically targeted in multiple cancer types. This form of therapy would be very beneficial to cancers with p53 aberrations.
7.2 BH3 mimetics
Directly targeting the BCL-2 family in cancers via BH3 mimetics has been an important milestone in modern chemotherapeutic strategies. These BH-3 mimetics act similarly to BH-3 derepressor/sensitiser BH-3 only proteins. Gossypol (AT101) is a BH3 mimetic molecule derived from the cottonseed plant. AT101 can interact with and inhibit the anti-apoptotic functions of BCL-2, BCL-xL, and MCL-1 (Elkholi et al., 2011), and potently induce apoptosis in many cancer cell lines.
AT101 has been shown to induce apoptosis in cisplatin-resistant ovarian cancer cells (Hu et al., 2012). In chemoresistant ovarian cancer cells BCL-2 and BCL-xL are overexpressed preventing BAX activation. AT101 not only induces BAX activation by binding to BCL-2 and BCL-xL (freeing BAX and other pro-apoptoic proteins), but also by upregulating PUMA and Noxa, and downregulating XIAPs, BCL-2, and BCL-xL. In cisplatin-resistant ovarian cancer cells AT101 induces via the Akt-p53 pathway, mitochondrial SMAC release. SMAC is the key mediator of apoptosis by AT101 in the chemoresistant cells. The mechanisms involved in the AT101-mediated release of SMAC and how it mediates apoptosis in these cancer cells remains to be elucidated. This example of AT101 highlights the need to fully understand the mechanisms involved in apoptosis and cancer treatments, so that we can treat and induce chemosensitivity in different cancer types.
ABT-737 is a BH3 mimetic that functions similarly to BAD. It can interact with and inhibit the anti-apoptotic function of BCL-2, BCL-w, and BCL-xL (Elkholi et al., 2011). The orally available form of ABT-737 is ABT-263. Tumours can become resistant to ABT-737 by expressing a high level of MCL-1. This concurs with the idea that tumour cells become dependent on anti-apoptotic proteins to sequester the anti-apoptotic proteins induced in oncogenesis.
7.3 BCL-2 dependence
In chronic lymphocytic leukaemia (CLL) cells are dependent on BCL-2 or MCL-1 to sequester the oncogenically induced pro-apoptotic proteins. As mentioned previously cells with high levels of MCL-1 are resistant to ABT-737. An assay called BH3 profiling can be used to predict dependence of cancers cells on anti-apoptotic proteins, and it can also predict cellular response to chemotherapy agents such as ABT-737 (Del Gaizo Mooreet al., 2007). BH3 profiling can also accurately predict between MCL-1 dependence and BCL-2 dependence in cancer cells.
BCL-2 dependence in CLL is indicative of high levels of BIM sequestered by BCL-2. These high levels of BIM concur with the idea that through oncogenic processes pro-apoptotic proteins can be induced, priming the cell for death, hence dependence on an anti-apoptotic protein to sequester the pro-apoptotic protein. In CLL with BCL-2dependence ABT-737 frees sequestered BIM from BCL-2, allowing BIM to directly activate BAX, resulting in MOMP (Del Gaizo Moore et al., 2007).
Human epidermal growth factor receptor (HER) 2 overexpressing breast cancers predominantly arise from cells that overexpress BCL-2, BCL-xL, or MCL-1. This dependence of anti-apoptotic proteins is required to sequester BIM which is expressed due to oncogenic signalling (Campone et al 2011). Overexpression of HER2 causes Akt signalling. Akt activates mammalian target of rapamycin complex 1 (mTORC1) through phosphorylation. Activated mTORC1 results in expression of the transcription factor c-Myc which occupies regions at the BIM promoter. This results in a cell that is primed for death, as there is a high number of BIM sequestered by the anti-apoptotic repertoire, and disrupting the sequestered BIM results in MOMP and apoptosis.
The mitotic inhibitor paclitaxel has been shown to induce apoptosis in breast cancer cells through a novel displacement mechanism in which BMF and PUMA displace BIM from bound anti-apoptotic proteins (Kutuk and Letai, 2010). This apoptotic function of paclitaxel is BIM dependent and so treatment would only work on cancer types with cells primed for death through BCL-2 dependence, or in conjunction with other drugs.
Herceptin is used to treat HER2 overexpressing breast cancers. It functions by interfering with the HER2 receptors, downregulating downstream oncogenesis. Cancer cells that gain resistance to herceptin have been shown to have an increase in BCL-2:BAX complexes, and these cells are chemosensitised by ABT-737 (Crawford and Nahta, 2011). Acquired resistance to chemotherapeutic agents is an important obstacle to overcome. Many acquired resistances arise through overexpression of anti-apoptotic proteins (e.g. BCL-2 dependence), and through targeting the BCL-2 family through BH-3 mimetics these chemoresistant cells can be chemosensitised. It is also important to understand the molecular mechanisms at work and how a cancer cell becomes resistant so it can be prevented or overcome by new novel therapies.
...
Most emerging novel cancer treatments function by downregulating specific targets. Future novel cancer treatments could target BCL-2 dependency by using peptides from the nuclear receptor protein Nur77 turning BCL-2 into a pro-apoptotic protein (Kolluri et al., 2008), or by inducing BIM with a diterpenoid derivative to convert BCL-2 into an active BAX-like molecule, creating large BCL-2 pores (Zhao et al., 2011). Other novel treatments for BCL-2 dependence could involve targeting pathways, such as targeting Sigma-1 receptors with inhibitory ligands, preventing upregulation of BCL-2 (Meunier and Hayashi, 2011), or by upregulating INrf2, leading to increased BCL-2 degradation (Niture and Jaiswal, 2011). These possible novel therapies highlight the many ways we can treat diseases linked to the BCL-2 family.
Antisense oligonucleotide suppression has been used to target the BCL-2 family. Oblimersen (Genasense) downregulates the expression of the anti-apoptotic protein BCL-2. Resistance has been shown to develop in regards to this therapy (Rubenstein et al., 2011). Thus, antisense oligonucleotides should be used in conjunction with other therapies for benefit of the patient.
8.0 Conclusion
The BCL-2 family is a large and complicated family that are involved in processes other than regulating apoptosis. When a cell undergoes apoptosis there are 100’s, maybe 1000’s of different protein interactions simultaneously occurring. It is no surprise that it will take many more years of research to fully understand these proteins. Bioinformatics will be a key tool in understanding the complicated interplay among these pro-apoptotic and anti-apoptotic proteins. Whereas some of the molecular mechanisms have been elucidated in respect to the BCL-2 family, some family members such as the pro-apoptotic protein BCL-2-related ovarian killer (BOK) remain out of the limelight of research. BOK has been shown to possibly predispose certain cancer cells to chemotherapeutic treatment (Rodriguez et al., 2006) implicating it as a possible important area for cancer research.
The BCL-2 family are implicated in a plethora of diseases other than cancer and understanding the molecular mechanisms involved in these diseases is paramount.
There is still much debate on the very basis of how MOMP occurs, whether it is direct activator-dependent or not. The literature on the subject has conflicting data on both the direct activation model and indirect activation model. Even though through liposome studies and NMR analysis, BH3 proteins such as BIM have been shown to directly interact with and activate BAX and BAK (Gavathiotis et al., 2008: Dai et al., 2011) much controversy still exists. The reason these controversies still exist is that both models are too simple to cover the complicated intricate interactions of the BCL-2 family. The embedded together model (Leber et al., 2007) is a step in the right direction to consolidate the two opposing models.
Part 1
Part 1.1-2
part 2
part 3
part 4
part 5
part 6
@RiskDebonair
Adventure Capitalist of the Future