
The buzzword with Fluorescence microscopy is high.
In vivo observation of cellular process is one of the beautiful side-effects of this technology yet is not the only reason for its choice. When choosing a microscopy technique two parameters are of utmost importance: Contrast and Resolution. In a picture Dynamic range is also important but in the case of microscopy, quantification is not done visually.
What is it?
Is a tool to help you look inside of cells, fixed or alive. Tremendously useful due to properties like its extremely high contrast. With the help of CCD cameras by virtue of their linear range, You can label specific components with high specificity in a cell and set up experiments with multiple colors simultaneously that are highly quantitive and precise.
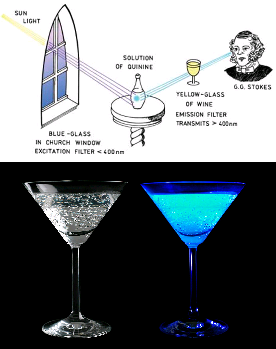
Many recorded accounts of observation have been made through history since the 16th, century. An example of this is Herschel's observation of the bark tree (containing quinine) phenomenon of blue light.
Tonic water (contains quinine), after excitation by UV light. A property regularly used by some bartenders
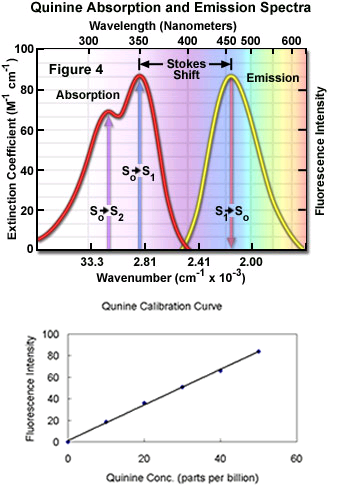
As a general rule, the excitation goes from High energy/Low wavelength to Low energy/High wavelength, with some exceptions. The difference between the maximum for excitation and the maximum for emission is called the Stokes shift.
What occurs during fluorescence can be seen as represented Jablonski diagram, used to represent the distance traveled by light during that time, the electrons in a molecule can absorb light and be excited, therefore going to a higher state of energy.
The whole lifetime of the fluorescence is in the order of nanoseconds. Absorption is relatively short-lived in the order of femtoseconds from S0 to S2 (To give an idea of how short these intervals are, light travels 0.3µm during the absorption time).
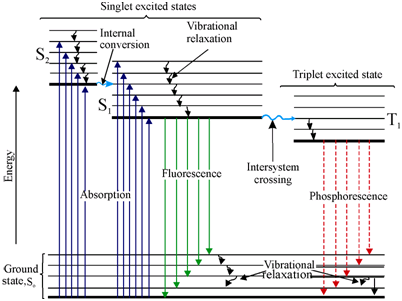
After the peak of excitement, it goes to the lowest level S2 to S1, the excited state this takes some picoseconds, is called internal conversion. It will stay in that stay in that time for a relatively long time, from 0.5 to 10 nanoseconds and on the way down a photon is emitted. This explains why normally the emitted photon has lower energy, due to the energy lost during internal conversion. If the process is slower the emission will take longer and this is called phosphorescence.
Separating the emission light from the fluorophore (color emitting molecule) light
Not always light will be emitted, as the energy can be transferred to other molecules before that happens or can lose energy through non-radiative decay, as energy can be disipated in the form of heat or movement. This is expressed as a ratio called quantum efficiency (QY). You want dyes with high QY (emitted photons/absorbed photons) and absorption coefficients.
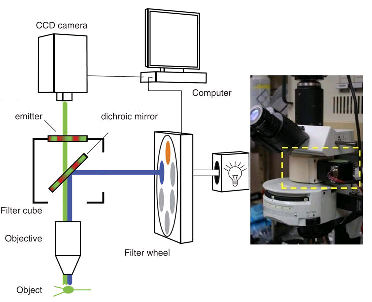
First, you need a light source to excite these molecules. You can use arc lamps, lasers or LEDs passed through a filter that only let pass the excitation light. The excitation light is reflected by the dichroic mirror (that only reflects light on certain wavelengths). This goes through the objective lens and the process of fluorescence occurs. Later the emitted fluorescence passes through the objective lens and goes to the dichroic mirror that is made so the fluorescence can pass through, and finally, there's an emission filter that stops any excitation light that has passed letting the fluorescence light go to the photoreceptor, be it a camera or retina.
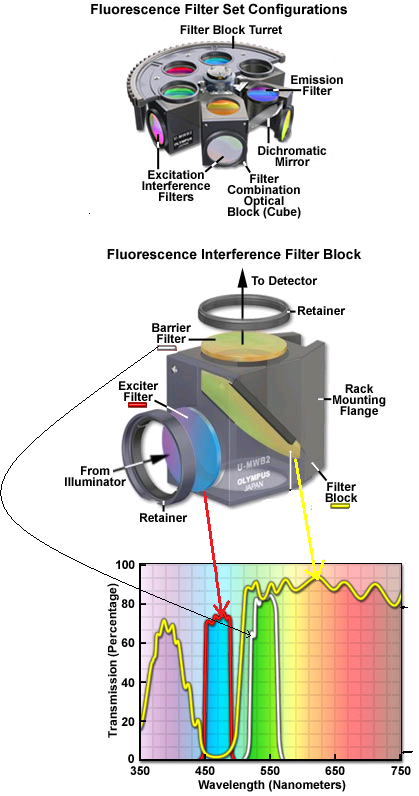
Normally all the equipment is combined in the shape of a cube (fluorescence interference filter block). So that in a single unit. You can have several of this cubes in arrangements to filter different types of fluorescence.
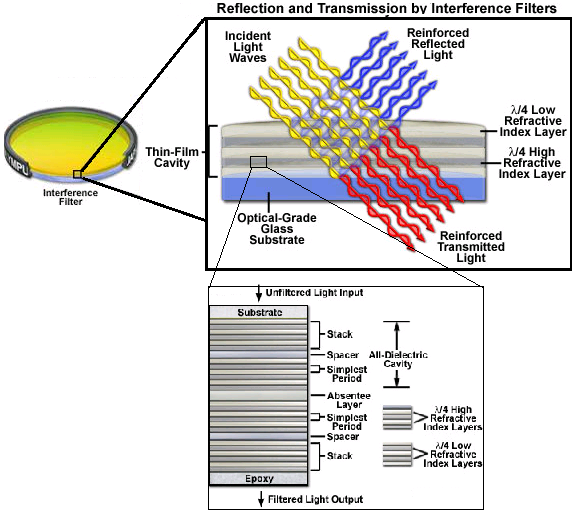
The interference filters have several layers, some are reflective, some are transparent. The distance between the layers is a half or a quarter of the wavelength of the light desired to allow to pass through. The incident light will bounce in between each one of the layers and with the help of constructive interference, the filtered light will pass to the next layer. So when the light doesn't match the distance between the layers the light will get destructive interference and will reflect.
The band of color after filtering will have to be selected carefully as normally the higher the wavelength the smaller the intensity of the non-filtered fluorescence. Also, the fluorophores can incur in photobleaching (their fluorescence fades too fast) due to molecule change or denaturation for instance due to oxygen (that take away energy by oxidizing the molecule and turns into reactive species that could destroy the fluorophore), for this one needs to select fade resistant dyes. Another approach is put as much dye as possible. Using additives like glycerol to counter oxygen exposition and budgeting with the shutter the time/intensity of light emission to only when acquiring data.
The classic organic dyes are coumarin (blue), fluorescein (green) Rhodamine (red). Fun side note: Fluorescein is the compound used to color the Chicago River during San Patrick's day.
Probes
The first synthesized fluorescent probe was fluorescein in 1871. Still in use to trace water flow underground. Its fluorescence is pH dependent, with fluorescence pH>7. In a Jablonski diagram, the energy difference between the HOMO (Highest occupied molecular orbital) and the LUMO (lowest unoccupied molecular orbital) is what determines the emission wavelength of the fluorophore (the smaller the difference, the redder the color)
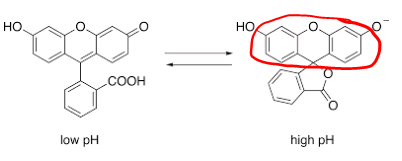
In the case of fluorescein, the part of the molecule that is important for its fluorescence is the extended conjugated structure created by the single and double bonds in the rings. Normally the longer the extended structure the lower the difference HOMO-LUMO and the redder the color. 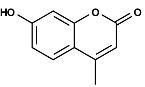 Coumarin has one less ring and its color is blue.
Coumarin has one less ring and its color is blue.
The rigidity in the structure stops the loss of energy by rotation. Another factor is the atomic composition of quantum effects. By tweaking this factors is possible for chemists to produce fluorophores that cover the whole visible spectrum. As a general rule, the lower wavelength fluorophores are better at capturing photons and are brighter.
Some of these dyes bind already to cellular components like DNA. Nonetheless, for the vast majority of uses, they need to be coupled with some kind of tracker.
In the case of proteins, there are two nucleophilic amino acids side chains that are used C and K (cysteine and lysine). They are only reactive in their deprotonated form or in high pH. In the case of cysteine as 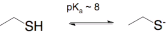 as thiolate >8
as thiolate >8
and in the case of lysine is reactive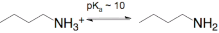 as a free amine >8 - 10.
as a free amine >8 - 10.
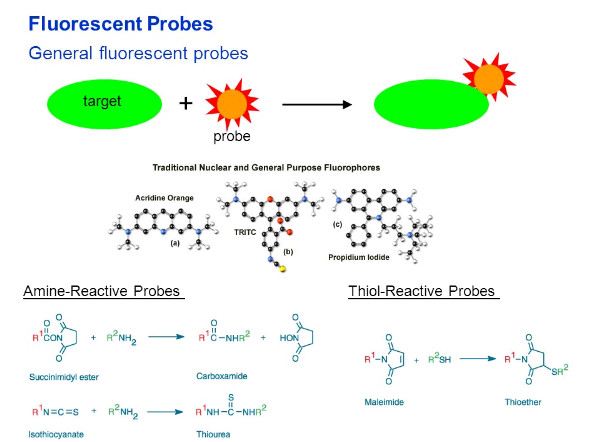
Not all the probes require covalently, an example of this is rhodamine-phalloidin for instance used to bind to actin and to some receptors.
There are nanocrystals made of semiconductors called quantum dots, requires layering with a biocompatible material such as a lipid or PEG. Their spectrum of fluorescence is dependent on their size, so the wavelength is size tuneable (from blue to red by increasing the size), but they respond to the same excitation light wave. They are a lot bigger and less specific around the size of a protein and their photobleaching is almost of no importance.
Proteins
First, it was done by labeling each target molecule, which was extremely laborious. Later Direct immunofluorescence allowed the use of specific targets since you labeled the antibody. Later it was found that using a labeled antibody to recognize a pair antibody-target was a lot easier and could give some amplification as more marked antibodies could attach to a single place, this is indirect immunofluorescence.
Another source is Fluorescent proteins. Of them, the most known are the GFP discovered in Jellyfish, a cofactor of aequorin already blue fluorescent. Discovered and isolated by Shimomura in 1962. The reaction to produce the chromophore requires oxygen and the most important part of the molecule is the highly conserved Gly67, present in all the fluorescent proteins. For every molecule of GFP, you get one Hydroxide peroxide.
An interesting thought is how Hemoglobin another chromophore, shifted to the red spectrum is related to oxygen.
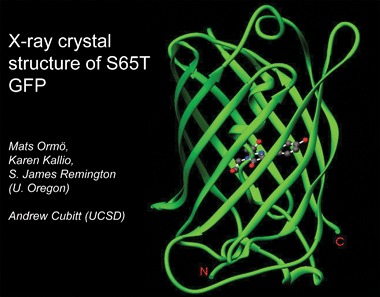
The wild-type isolated from jellyfish is not strongly fluorescent. Its emission peak glows best under UV. The produced mutants at S65 are the ones that have the restricted green spectrum that is known for, the reason the more used is the eGFP (e stands for enhanced) that is modified to fold at higher temperatures like the ones that mammalian cells have.
It can be seen in the structure the 11-stranded beta-barrel with a single internal alpha helix bearing the chromophore.
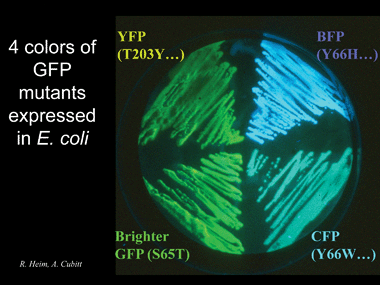
One of the problems with GFP is that it tends to dimerize and if united to a target molecule it could create a dimer of the target protein. A problem that has been eliminated for the most part. Other colors can be obtained by mutations.
After that people started looking for homologs and discovered that they are widely distributed in nature. In fact in Bilateria, vertebrates like us there are homologs but they are not fluorescent. Most of them come from Cnidaria, that includes jellyfish and corals.
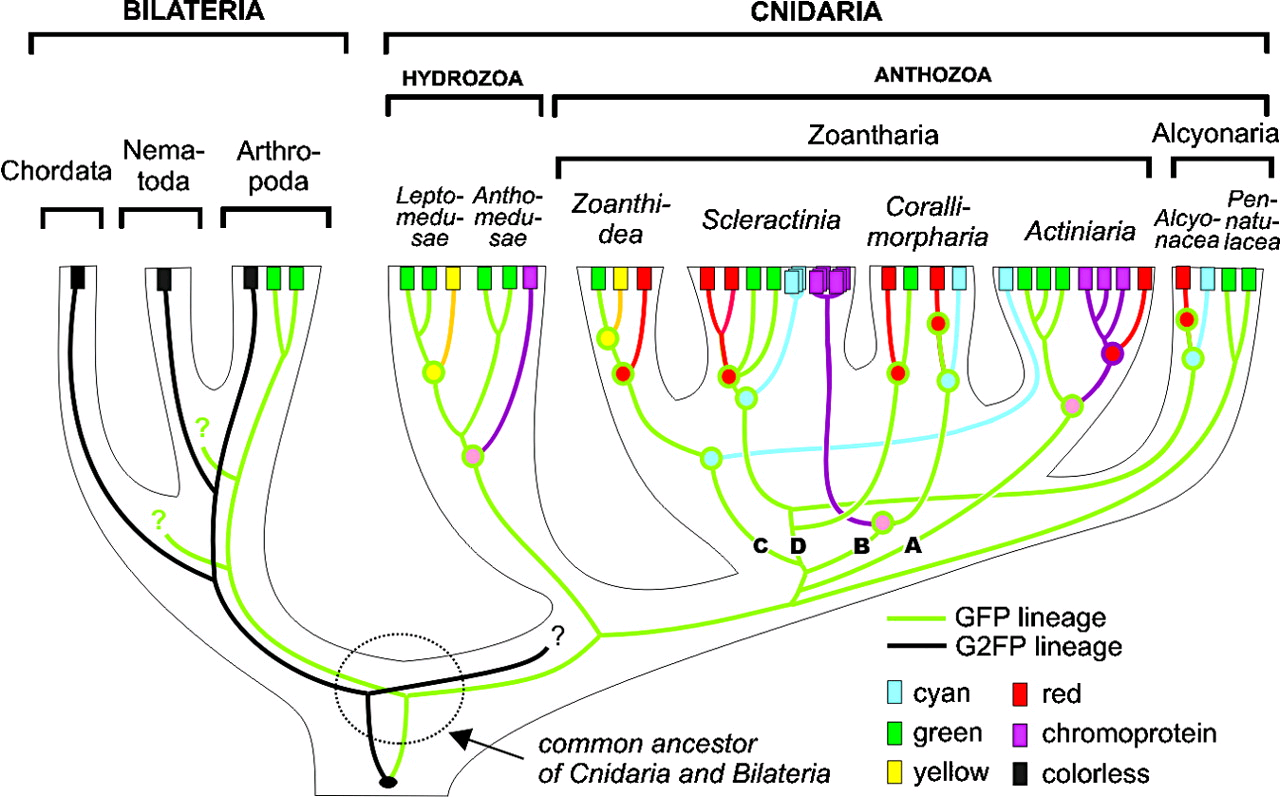
The discovery of Corals as having homologs allowed to produced mutants with a wide variety of colors. The initial structure like DsRed was tetrameric and hard to manipulate but today there are many derivates.
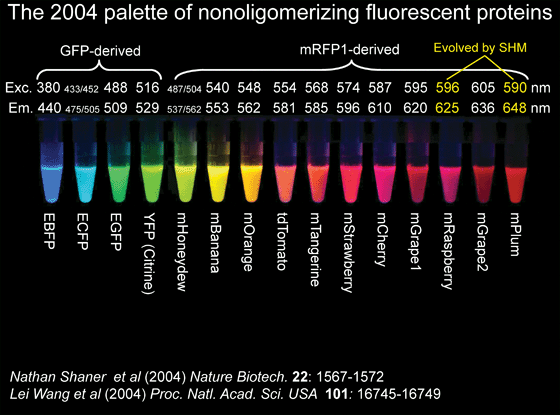
Now there are even proteins that are photoactivable, which mean they fluoresce in a spectrum and when they absorb a different wavelength they change. Like is the case with "Dendra" that emits green light and turns red when blue light shines on it. or "Dronpa" a reversible protein fluorochrome. When the blue light is emitted on from the green color starts to fade out until is almost gone, but when the blue light is taken away it comes back.
In the case of mammals study, the in-vivo use of fluorescent proteins poses a problem as mammals are opaque bellow 600nm, due to the peaks of emission of hemoglobin. Example of this is the principle of transillumination, used in medical diagnosis.
[Image source: Link]
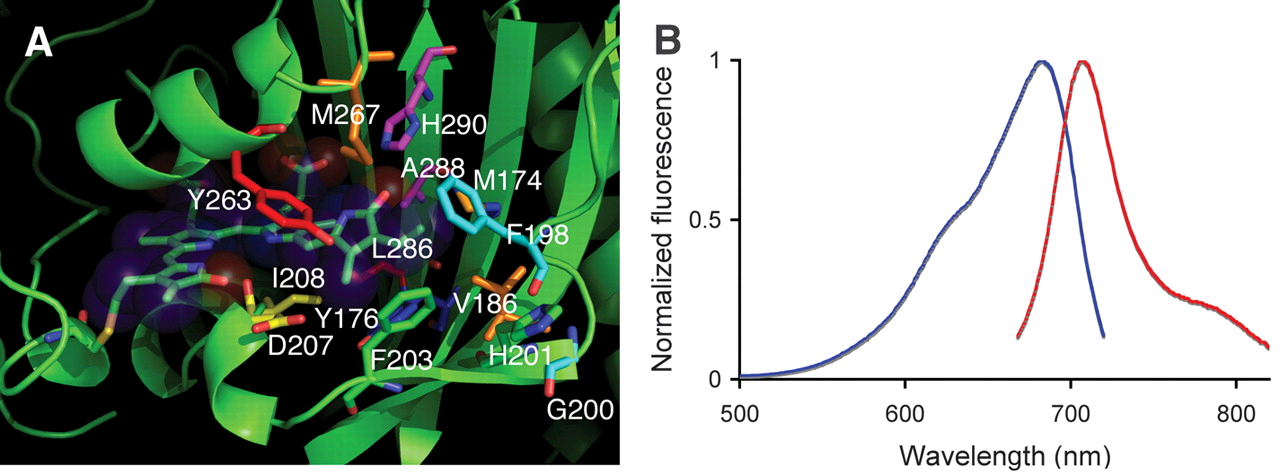
Emission becomes weaker as it goes higher in wavelength with previous fluorochromes. So a new family of fluorochromes derived from heme synthesis was developed in the form of tetrapyrrole pigments. Biliverdin a product of degradation of heme, widely known for its role in bruises, is almost ubiquitous and is used by some bacteria to sense light. This allowed the creation of mutants that can emit >700nm in the infrared range.
References
